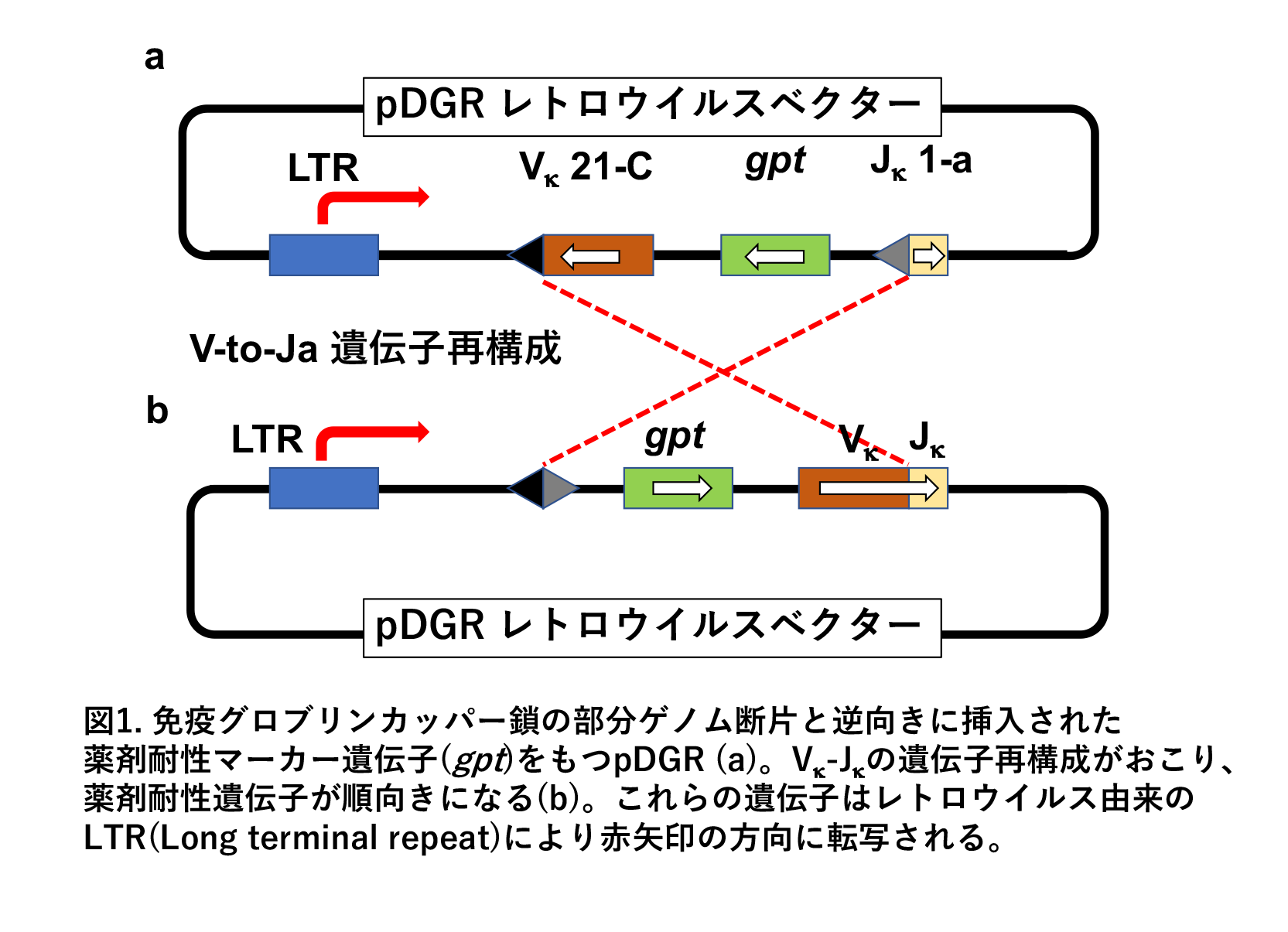
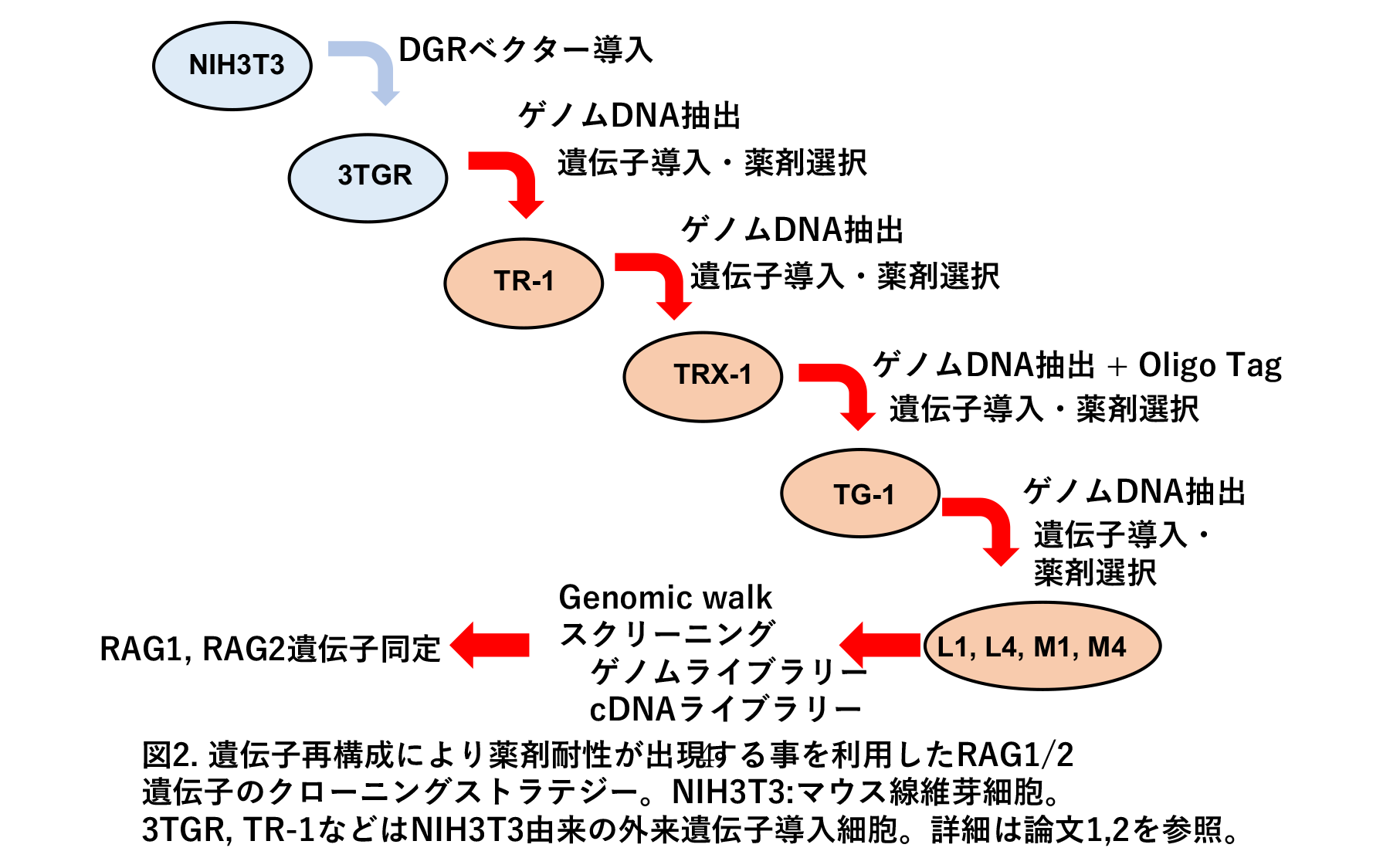
This post reflects on the long and winding road that led to the discovery of the
oxidative stress-inducible gene, interleukin 11, as a marker for cancerassociated
fibroblasts.
Hiroyasu Nakano and Takashi Nishina
It has been over ten years since we started a project to visualize oxidative stress
in vivo with a murine model. To achieve this goal, we first hypothesized that
we needed to identify the gene (s) induced by oxidative stress. Once we
identified the responsible gene(s), we constructed vectors that harboured a
fluorescent protein gene, such as enhanced green fluorescence protein (Egfp), placed
under the control of the promoter of oxidative stress-induced genes. The
promoter of those genes could be then used to control EGFP expression to
visualize oxidative stress. Based on this strategy, we first attempted to identify
oxidative stress-inducible genes by stimulating murine embryonic fibroblasts
with hydrogen peroxide in a microarray analysis. The expression levels of over
200 genes rapidly increased more than 4-fold. We first selected 10 genes from
the top of the list of upregulated genes, which showed more than 30-fold
increases in expression. Considering that our final goal was to visualize
oxidative stress in vivo, genes that were upregulated by oxidative stress in
vivo were considered candidate genes.
We previously reported that oxidative stress was tightly associated with cell
death1; therefore, we surmised that a murine acute liver injury model might be
suitable for testing whether these candidate genes were upregulated. Injecting
mice with anti-Fas antibody was known to induce acute liver injury2. To our
surprise, after injecting the anti-Fas antibody into mice, and analysing the
livers, we found that the ten candidate genes did not show any increase in
expression. Thus, we needed to examine 200 other genes that were upregulated
more than 4-fold after oxidative stress. For this analysis, we needed to come up
with an efficient method for selecting candidate gene(s) for vector construction.
After struggling on this project for several months, a candidate gene emerged
from another project in my lab, during an analysis of livers from Cflarflox/flox;Mx1-
Cre mice. By using tissue specific Cflar-deficient mice3, we investigated a role of
anti-apoptotic protein, cellular FLICE-inhibitory protein that is encoded
by Cflar gene. The interferon (IFN)-inducible Mx1-Cre/loxP system was a
prototype of the inducible Cre recombinase system, where a gene flanked by
two loxP sites could be inactivated. We injected Cflarflox/flox;Mx1-Cre mice with
polyinosinic:polycytidylic acid (poly I:C) that induces the production of
IFNα/β by Kupffer cells. This procedure resulted in the expression
of Cre recombinase in hepatocytes. We found that a Cre-dependent deletion of
the Cflar gene in hepatocytes induced fulminant hepatitis4.
A microarray analysis revealed that, following the poly I:C injection, the most
upregulated gene in the livers of Cflarflox/flox;Mx1-Cre mice was interleukin 11
(Il11), which was unfamiliar to us at that time. No one in the lab, including me,
had paid attention to IL-11 before. According to my lab notes, on the night of
February 28, 2009, I happened to find that Il11 was also on the list of
upregulated genes in fibroblasts in response to oxidative stress! This indicated
that IL-11 might be a potential candidate for visualizing oxidative stress in vivo.
After performing many experiments to verify that IL-11 was indeed induced by
oxidative stress in vitro and in vivo, we finally decided to generate Il11-
Egfp reporter mice.
To generate Il11-Egfp reporter mice, we used a knock-in strategy to insert
the Egfp gene just downstream of the ATG initiation site of the Il11 gene in a
bacterial artificial chromosome (BAC). We initiated the generation of Il11-
Egfp BAC transgenic mice in collaboration with Professor Otsuka at Tokai
University, and Dr. Tada at Juntendo University, in 2010. We assumed that it
would take several months to generate Il11-Egfp BAC transgenic mice. A
talented scientist, Takashi Nishina, the first author of the present paper5, had
recently joined our lab as a postdoctoral fellow in April, 2010. Before working
on Il11-Egfp reporter mice, we investigated the role of IL-11 with an
acetaminophen (APAP)-induced liver injury model. That study took almost two
years to complete6. Then, Takashi concentrated his efforts on analysing Il11-
Egfp reporter mice. We first examined whether Il11-Egfp mice could be used to
monitor IL-11-producing cells in an APAP-induced liver injury model.
However, we could not detect any EGFP signals in the liver. Those results were
3
disappointing, but Takashi did not give up and continued focusing on Il11-
Egfp reporter mice.
Around that same time, an Australian group published a seminal paper on IL-
11. They reported that IL-11 expression was elevated in gastric and colorectal
cancer (CRC) in human patients and in several murine models7. Moreover, they
also showed that a blockade of IL-11 signalling, in IL-11 receptor knockout mice
or with an IL-11 antagonist, resulted in a dramatic reduction in CRC
development7. Given that oxidative stress is tightly associated with the
development of various cancers8, and that IL-11 was induced by oxidative
stress6, we hypothesized that IL-11 might be the missing link between oxidative
stress and tumorigenesis. Thus, Takashi induced colitis in mice with dextran
sodium sulphate (DSS) to visualize IL-11+ fibroblasts in vivo. Following DSS
treatment, we successfully detected IL-11+ (EGFP+) cells in mouse colons, but
the percentage of IL-11+ cells was very low. Nevertheless, Takashi isolated IL-
11+ cells from those mice and characterized their gene expression profiles in a
microarray analysis. Most IL-11+ cells appeared to express stromal cell markers,
such as Thy1, podoplanin, and Sca1; thus, they were considered to be stromal
fibroblasts.
In April 2014, our group moved to Toho University, where we continued and
expanded our work on IL-11. We investigated the role of IL-11 further by
generating Il11-/- mice and anti-IL-11 antibody9. We could visualize IL-11+ cells
in tumour tissues in the colons of mice treated with azoxymethane/DSS and in
the progeny of ApcMin/+ mice crossed with Il11-Egfp mice. Of note, IL-
11+ fibroblasts were located adjacent to tumour cells (Figure 1).
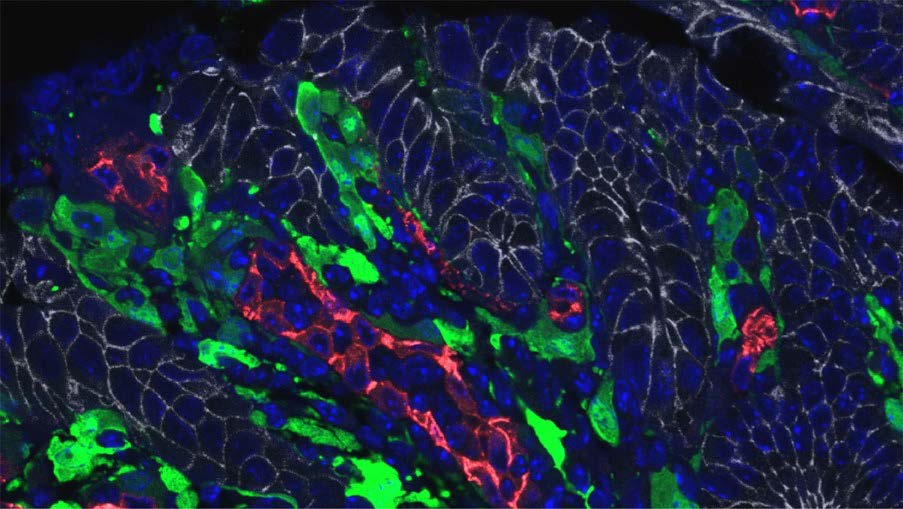
Figure 1. IL-11+ cells are located adjacent to tumor cells. Colon tumor sections
from ApcMin/+;Il11-Egfp mice were stained with anti-GFP (IL-11+ cells, green),
anti-E-cadherin (tumor cells, white), anti-CD31 (endothelial cells, red), and
DAPI (nuclei).
We combined these results with a meta-analysis of human cancer databases in
an article published in April, 20215. However, that was not the end of the IL-11
story. Our present study revealed a correlation between IL-11+ fibroblasts and
the development of CRC. Therefore, our next goal is to show directly that IL-
11+ fibroblasts contribute to the development of CRC, with another approach.
For that purpose, we conceived a strategy for depleting IL-11+ fibroblasts after
the development of CRC. If that strategy works well, we can conclude that IL-
11+ fibroblasts are indeed cancer-promoting fibroblasts, and they might serve as
new targets for treating CRC.
References
- 1.Nakano H, Nakajima A, Sakon-Komazawa S, Piao JH, Xue X, Okumura K.
Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell
Death Differ 13, 730-737 (2006).
- 2.Ogasawara J, et al. Lethal effect of the anti-Fas antibody in
mice. Nature 364, 806-809 (1993).
- 3.Nakano H, Piao X, Shindo R, Komazawa-Sakon S. Cellular FLICEInhibitory
Protein Regulates Tissue Homeostasis. Curr Top Microbiol
Immunol 403, 119-141 (2017).
- 4.Piao X, et al. c-FLIP maintains tissue homeostasis by preventing apoptosis
and programmed necrosis. Sci Signal 5, ra93 (2012).
- 5.Nishina T, et al. Interleukin-11-expressing fibroblasts have a unique gene
signature correlated with poor prognosis of colorectal cancer. Nat Commun 12,
2281 (2021).
- 6.Nishina T, et al. Interleukin-11 links oxidative stress and compensatory
proliferation. Sci Signal 5, ra5 (2012).
- 7.Putoczki TL, et al. Interleukin-11 is the dominant IL-6 family cytokine
during gastrointestinal tumorigenesis and can be targeted
therapeutically. Cancer Cell 24, 257-271 (2013).
- 8.Hayes JD, Dinkova-Kostova AT, Tew KD. Oxidative Stress in
Cancer. Cancer Cell 38, 167-197 (2020).
- 9.Deguchi Y, et al. Generation of and characterization of anti-IL-11
antibodies using newly established Il11-deficient mice. Biochem Biophys Res
Commun 505, 453-459 (2018).
(BEHIND THE PAPER; April 16, 2021.
https://cancercommunity.nature.com/posts/interleukin-11-expressing-fibroblasts-have-aunique-
gene-signature-correlated-with-poor-prognosis-of-colorectal-cancer)